Pheatmap in R: Erstellen von anpassbaren gruppierten Heatmaps
- Name
- Sebastian Brandt
Published on
Heatmaps sind ein unverzichtbares Werkzeug im Werkzeugkasten eines Datenwissenschaftlers und stellen eine visuell intuitive Darstellung komplexer Datensätze dar. Unter den verschiedenen verfügbaren Paketen in R zur Erzeugung von Heatmaps sticht Pheatmap durch seine Flexibilität und Anpassungsoptionen hervor. Dieser Artikel führt Sie durch den Prozess der Erstellung von schönen, anpassbaren gruppierten Heatmaps mit Pheatmap in R.
Pheatmap ist mehr als nur eine Funktion in R; es ist ein leistungsstarkes Werkzeug, das den Benutzern ermöglicht, Heatmaps mit größerer Kontrolle und Anpassungsoptionen als die Standard-R-Heatmap-Funktion zu erstellen. Mit Pheatmap können Benutzer die Visualisierung der Genexpressionsanalyse, die Zeichnung von Korrelations-Heatmaps und die Anpassung von Labelgrößen und Dendrogramm-Sichtbarkeit visualisieren. Tauchen wir ein in die Welt von Pheatmap und erkunden seine Fähigkeiten.
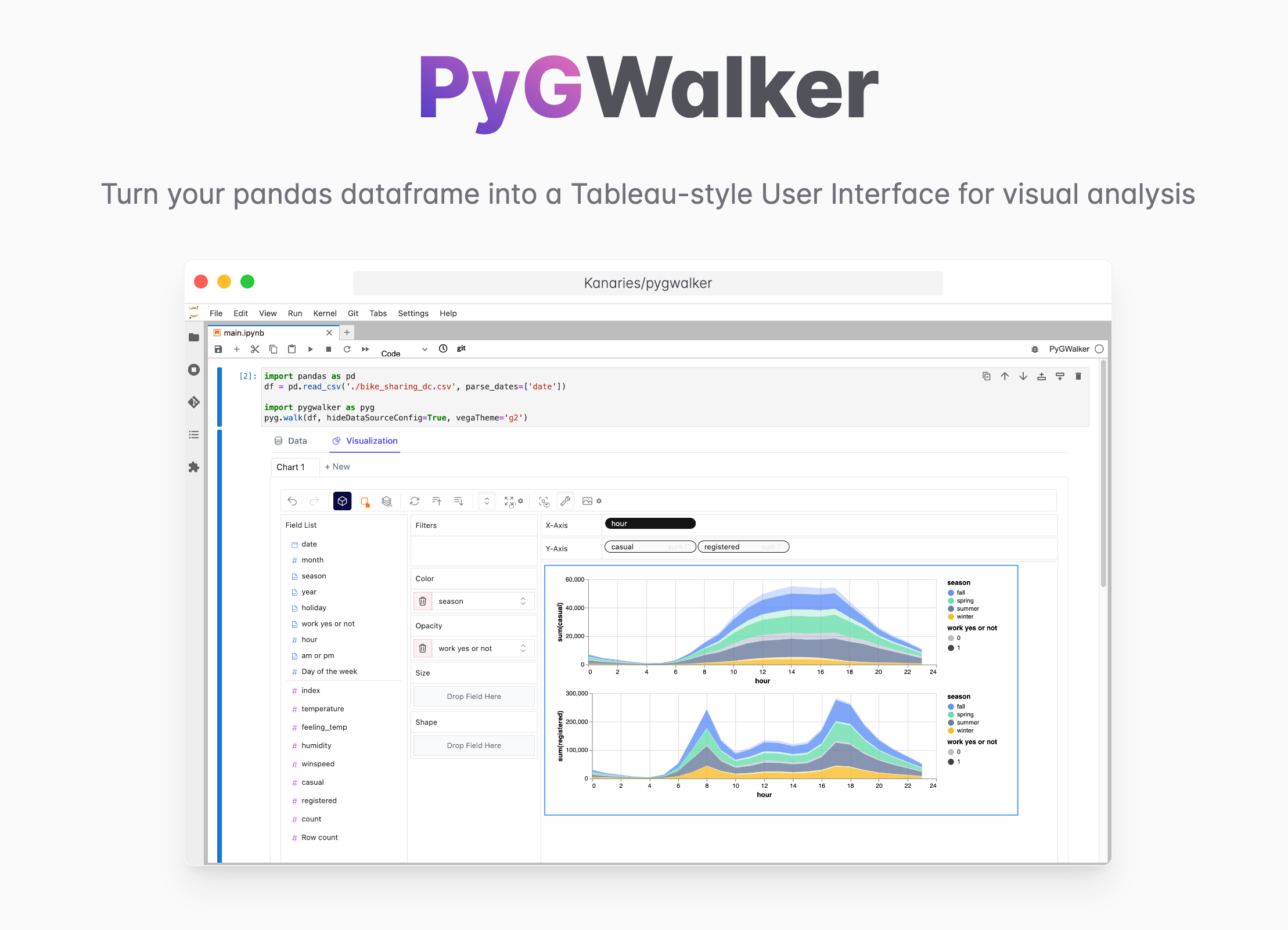
Möchten Sie schnell Datenvisualisierung aus Python Pandas DataFrame ohne Code erstellen?
PyGWalker ist eine Python-Bibliothek für explorative Datenanalyse mit Visualisierung. PyGWalker (opens in a new tab) kann Ihren Jupyter Notebook-Datenanalyse- und Datenvisualisierungs-Workflow vereinfachen, indem es Ihr Pandas DataFrame (und Polars DataFrame) in eine Tableau-ähnliche Benutzeroberfläche für die visuelle Exploration umwandelt.
Was ist Pheatmap in R?
Pheatmap ist eine Funktion in R, die schöne Heatmaps generiert und es Datenwissenschaftlern ermöglicht, komplexe Daten auf vereinfachte Weise zu visualisieren. Es bietet mehr Kontrolle und Anpassungsoptionen im Vergleich zu den Standard-R-Heatmap-Funktionen wie heatmap() und heatmap.2(). Pheatmap zeichnet sich durch seine Fähigkeit aus, ästhetisch ansprechende und informative Heatmaps zu erstellen.
Pheatmap ist besonders nützlich in der Genomik, wo es häufig zur Visualisierung von Genexpressionsdaten verwendet wird. Es ermöglicht die Hinzufügung von Annotationen und verwendet Clustering-Methoden, um ähnliche Daten zu gruppieren, was die Interpretierbarkeit der Heatmap erhöht. Es bietet auch Optionen zur Zeilen-/Spalten-Z-Score-Standardisierung, die in bestimmten Datenanalyse-Szenarien entscheidend sein können.
Wie funktioniert Pheatmap?
Pheatmap funktioniert, indem es eine Matrix von Daten nimmt und sie in eine visuell intuitive Heatmap umwandelt. Die Datenwerte werden als Farben in der Heatmap dargestellt, wobei die Farbintensität die Größe des Werts angibt. Dies ermöglicht eine einfache Identifizierung von Mustern und Korrelationen in den Daten.
Die Funktion führt auch eine hierarchische Clusteranalyse der Daten durch, indem sie ähnliche Zeilen und Spalten zusammen gruppiert. Dies wird visuell durch ein Dendrogramm dargestellt, ein baumartiges Diagramm, das die hierarchische Beziehung zwischen den Datenpunkten zeigt. Die von Pheatmap verwendete Clustering-Methode kann je nach Bedarf des Benutzers angepasst werden.
Pheatmap ermöglicht auch eine hohe Anpassungsfähigkeit des Aussehens der Heatmap. Benutzer können die Farbpalette, die Labelgrößen, die Sichtbarkeit des Dendrogramms und mehr kontrollieren. Dies macht Pheatmap zu einem vielseitigen Tool für die Datenvisualisierung in R.
Vorteile von Pheatmap gegenüber der Standard-R-Heatmap
Während die Standard-R-Heatmap-Funktion nützlich ist für die grundlegende Erzeugung von Heatmaps bietet Pheatmap mehrere Vorteile, die es zu einer bevorzugten Wahl für viele Datenwissenschaftler machen.
Erstens bietet Pheatmap mehr Kontrolle über das Aussehen der Heatmap. Benutzer können die Farbpalette anpassen, die Labelgrößen anpassen und die Sichtbarkeit des Dendrogramms kontrollieren. Dies ermöglicht die Erstellung von Heatmaps, die nicht nur informativ, sondern auch visuell ansprechend sind.
Zweitens führt Pheatmap eine hierarchische Clusteranalyse der Daten durch, indem ähnliche Zeilen und Spalten zusammen gruppiert werden. Dies erhöht die Interpretierbarkeit der Heatmap und ermöglicht eine einfachere Identifizierung von Mustern in den Daten.
Drittens ermöglicht Pheatmap die Hinzufügung von Annotationen und die Verwendung von Filtern, was besonders nützlich bei der Genexpressionsanalyse sein kann. Es bietet auch Optionen zur Zeilen-/Spalten-Z-Score-Standardisierung, die mehr Flexibilität in der Datenanalyse bieten.
Zusammenfassend lässt sich sagen, dass die Standard-R-Heatmap-Funktion ein nützliches Werkzeug für die grundlegende Erzeugung von Heatmaps ist, Pheatmap jedoch eine höhere Kontrolle und Anpassbarkeit bietet, was es zu einem leistungsstarken Tool für die Datenvisualisierung in R macht.
Anpassen des Aussehens von Pheatmap in R
Einer der Hauptvorteile von Pheatmap ist die Möglichkeit, das Aussehen der Heatmap an Ihre spezifischen Bedürfnisse anzupassen. So können Sie es tun:
Farbanpassung
Pheatmap ermöglicht es Ihnen, die Farbpalette anzupassen, die in der Heatmap verwendet wird. Dies kann mit dem Parameter color in der Funktion pheatmap() erfolgen. Sie können aus einer Vielzahl von Farbpaletten wählen, die in R verfügbar sind, oder Ihre eigene erstellen.
Label-Anpassung
Die Größe und das Aussehen der Labels in der Heatmap können mit den Parametern fontsize und fontface angepasst werden. Dies ermöglicht es Ihnen, die Lesbarkeit der Heatmap zu kontrollieren und sie entsprechend Ihren Präsentationsbedürfnissen anzupassen.
Dendrogramm-Sichtbarkeit
Pheatmap ermöglicht es Ihnen, die Sichtbarkeit des Dendrogramms zu kontrollieren, einem baumartigen Diagramm, das die hierarchische Beziehung zwischen den Datenpunkten zeigt. Dies kann mit den Parametern show_rownames und show_colnames in der Funktion pheatmap() erfolgen.
Hinzufügen von Annotationen
Pheatmap ermöglicht es Ihnen, Annotationen zur Heatmap hinzuzufügen, was bei der Genexpressionsanalyse besonders nützlich sein kann. Dies kann mit den Parametern annotation_row und annotation_col in der Funktion pheatmap() erfolgen.
Zusammenfassend bietet Pheatmap ein hohes Maß an Anpassungsmöglichkeiten, um Heatmaps zu erstellen, die nicht nur informativ, sondern auch visuell ansprechend sind. Egal ob Sie genomische Daten visualisieren oder Korrelations-Heatmaps erstellen, Pheatmap bietet die Flexibilität und Kontrolle, die Sie benötigen, um in R schöne, anpassbare Heatmaps zu erstellen.
Verwendung der Cluster-Methode durch Pheatmap
Pheatmap verwendet hierarchisches Clustering, um ähnliche Datenpunkte zusammenzufassen. Dies ist eine Methode der Clusteranalyse, die versucht, eine Hierarchie von Clustern aufzubauen. Das Ergebnis ist eine baumartige Darstellung der Daten, die als Dendrogramm bezeichnet wird. Dies ermöglicht es den Benutzern, die Daten auf eine Weise zu visualisieren, die die Beziehungen zwischen den Datenpunkten hervorhebt.
In Pheatmap kann die Cluster-Methode mithilfe der Parameter clustering_distance_rows und clustering_distance_cols für Zeilen bzw. Spalten angepasst werden. Die Standardmethode ist "euclidean", aber es können auch andere Methoden wie "maximum", "manhattan", "canberra", "binary" oder "minkowski" verwendet werden.
Erstellen von Heatmaps in R mit Pheatmap
Das Erstellen einer Heatmap mit Pheatmap in R ist einfach. Hier ist ein einfaches Beispiel:
# Lade die pheatmap Bibliothek
library(pheatmap)
# Erstelle eine Matrix von Daten
data <- matrix(rnorm(200), 20, 10)
# Generiere die Heatmap
pheatmap(data)Dies erzeugt eine grundlegende Heatmap mit den Standard-Einstellungen. Sie können die Heatmap anpassen, indem Sie Parameter zur pheatmap() Funktion hinzufügen. Zum Beispiel können Sie mithilfe des Parameters color die Farbpalette ändern:
# Definiere eine Farbpalette
my_palette <- colorRampPalette(c("blau", "weiß", "rot"))(25)
# Generiere die Heatmap mit der benutzerdefinierten Farbpalette
pheatmap(data, color = my_palette)Anpassung von Farben in Pheatmap
Pheatmap ermöglicht eine hohe Farbanpassung. Sie können Ihre eigene Farbpalette definieren und auf die Heatmap anwenden. Dies wird mithilfe des color Parameters in der pheatmap() Funktion gemacht. Hier ist ein Beispiel:
# Definiere eine Farbpalette
my_palette <- colorRampPalette(c("blau", "weiß", "rot"))(25)
# Generiere die Heatmap mit der benutzerdefinierten Farbpalette
pheatmap(data, color = my_palette)In diesem Beispiel wird die Funktion colorRampPalette() verwendet, um eine Palette von 25 Farben von Blau über Weiß bis Rot zu erstellen. Diese Palette wird dann mithilfe des color Parameters auf die Heatmap angewendet.
Zusammenfassung
Zusammenfassend ist Pheatmap ein leistungsstolles Werkzeug zur Erstellung anpassbarer clusterter Heatmaps in R. Egal ob Sie genomische Daten visualisieren, Korrelations-Heatmaps erstellen oder einfach nur Ihre Daten erkunden möchten, Pheatmap bietet die Flexibilität und Kontrolle, die Sie benötigen.
Häufig gestellte Fragen
Was sind die Vorteile der Verwendung von Pheatmap gegenüber der Standardbasis-R-Heatmap?
Pheatmap bietet mehrere Vorteile gegenüber der Standardbasis-R-Heatmap-Funktion. Es bietet mehr Kontrolle über das Erscheinungsbild der Heatmap, führt hierarchisches Clustering auf den Daten aus und ermöglicht das Hinzufügen von Annotationen und die Verwendung von Filtern. Dies macht es zu einem leistungsstarken Werkzeug für die Datenvisualisierung in R.
Wie kann ich die Farbpalette in Pheatmap anpassen?
Sie können die Farbpalette in Pheatmap mithilfe des color Parameters in der pheatmap() Funktion anpassen. Sie können aus einer Vielzahl von Farbpaletten wählen, die in R verfügbar sind, oder Ihre eigene erstellen.
Welche Clustering-Methoden verwendet Pheatmap?
Pheatmap verwendet hierarchisches Clustering, um ähnliche Datenpunkte zusammenzufassen. Die Cluster-Methode kann mit den Parametern clustering_distance_rows und clustering_distance_cols angepasst werden. Die Standardmethode ist "euclidean", aber andere Methoden wie "maximum", "manhattan", "canberra", "binary" oder "minkowski" können ebenfalls verwendet werden.